Omni-C
A proximity-ligation protocol using a sequence-independent endonuclease, generating data for TAD identification and scaffolding [1].
The Omni-C® kit from Dovetail® produces high-quality proximity ligation libraries. This protocol increases the genomic coverage and reduces biases when compared with restriction enzyme-based Hi-C preps. This protocol is used for identification of topologically associated domains (TADs), or when even coverage across the genome is needed, for example for SNP calling and genotyping, it is also an efficient scaffolding method. No extraction is needed, as the prep starts directly from cells or finely ground tissue.
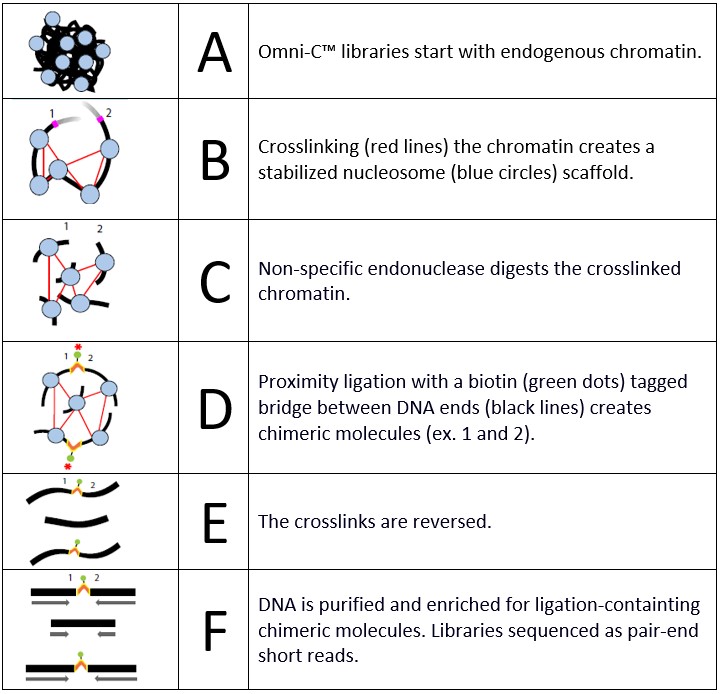
The library preparation consists of 5 stages:
- Sample preparation and cross-linking
Chromatin is fixed in place using formaldehyde. After cross-linking is stopped, an in situ digestion of the chromatin is performed using an endonuclease enzyme. - Lysate quantification
Chromatin is released by lysing the cells. In this step, the amount of chromatin obtained as well as the degree of digestion is assessed to ensure the success of the library prep. - Proximity ligation
End-polishing, ligation of a biotinylated oligonucleotide bridge. Intra-aggregate ligation to capture contacts is performed, followed by cross-link reversal and DNA purification. - Library preparation
End repair, adapter ligation, and purification steps result in the template for the final stage. - Ligation capture and amplification
A streptavidin enrichment step allows the capture of products from the proximity ligation step. Indices are added by PCR and a bead purification and size selection yield Illumina-compatible libraries. Up to 24 libraries can be multiplexed.
The expected result is that the sequenced libraries will contain information about which parts of the genome were physically proximal in the nuclei of the cells or tissue used to generate the library. This information can be used to build contact maps of the samples after aligning the reads to the reference genome or to scaffold a genome assembly into chromosome or near chromosome-scale scaffolds.
Regardless of sample type, Omni-C should generate libraries with high complexity and long-range information, even at low input. When compared to other restriction enzyme-based Hi-C data, we expect to have a more evenly distributed coverage across the genome. This allows for down-stream analyses such as SNP calling and phasing.
The even coverage of the Omni-C data should also make the scaffolding of contigs for genome assembly more accurate since no contig will be missing due to the lack of restriction sites, as may happen with classic Hi-C.
[1] https://ngisweden.scilifelab.se/methods/dovetail-omni-c/
[2] https://dovetailgenomics.com/wp-content/uploads/2021/09/Omni-C-Tech-Note.pdf