The sequencing of the whole genome
The approach used to study the genome of an organism differs depending on the sequencing challenge.
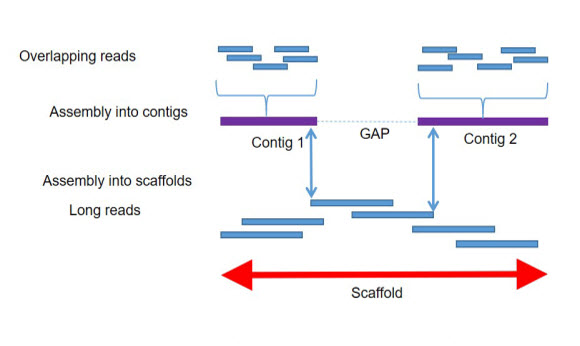
De novo sequencing enables the reconstruction of an unknown genome, not referenced in databases through the assembly of sequence data. Bioinformatics tools use sequence overlaps to build contigs of the longest possible size. The contiguous ones are themselves assembled together to create “scaffolds” and thus generate an alignment of sequences along the entire length of the genome. Depending on the technology used, it is necessary to combine sequencing with short and long reads for more precision in the assembly (Fig1).
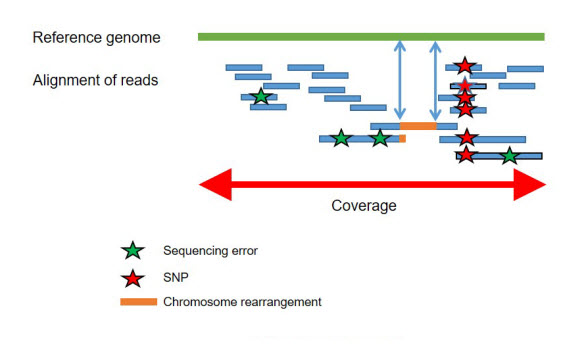
Resequencing (WGS) of already known genomes has a major challenge to identify nucleotide and structural variations and to understand their biological consequences. The short sequences resulting from resequencing are aligned with the reference sequence to identify genomic variations like SNPs. The combination of long reads with short reads sequencing is recommended for the accurate detection of CNVs, indels and chromosomal rearrangements (Fig2).
“Paired end” sequencing consists of sequencing both ends of short fragments, smaller than 1kb in size (short reads).
Several techniques are available to produce so-called long reads sequencing:
– the production of “Mate Pair” libraries allows to sequence both ends of fragments of several kilobases (kb) to be sequenced.
– PacBio “SMRT” technology (RSII, Sequel) can produce long reads sequencing (>20kb), and theoretically obtain a sufficient coverage for a de novo assembly, but its high cost hampers its use for the large eukaryotic genomes.
– the synthetic long reads technology is based on a molecular indexing system of long fragments (up to 100kb) allowing short reads from these large fragments to be physically linked together. This strategy is proposed by 10X Genomics technology (GemCode and Chromium) to facilitate assembly.
– the nanopore technology from Oxford Nanopore Technology can generate very long reads sequences (>2Mb), but still produces a high error rate that can be corrected by combining it with more accurate short reads sequencing.
– the optical mapping combined with the paired end sequencing enables to carry out a precise assembly, particularly for microbial genomes.