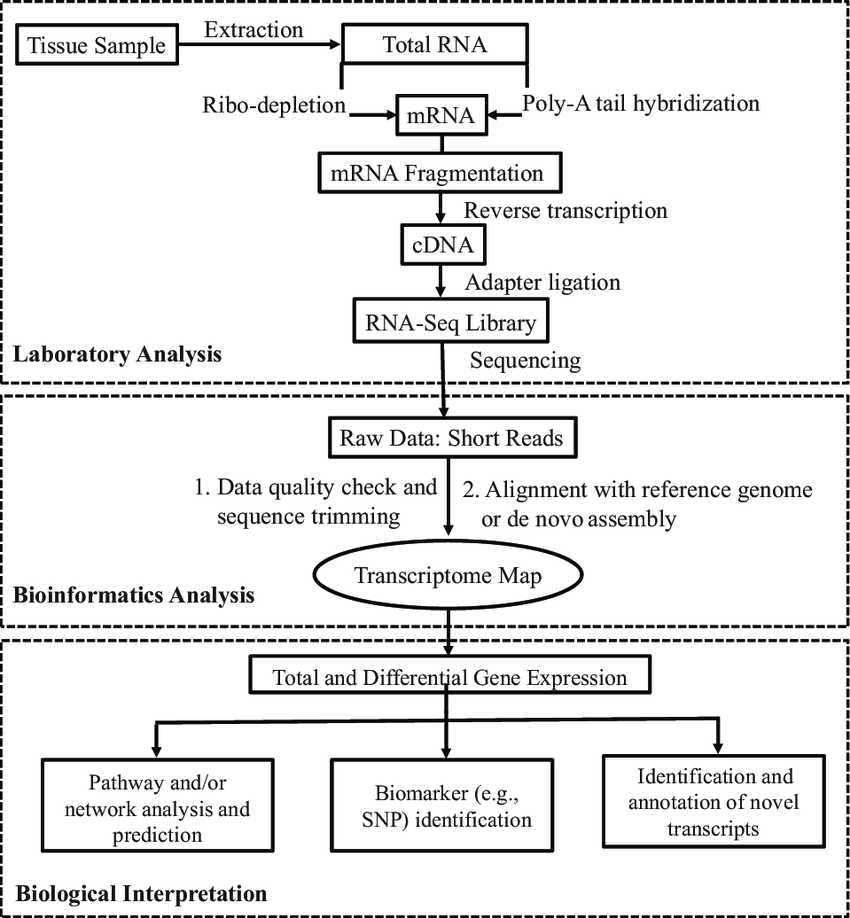
RNA‑Seq sequencing
The high-throughput RNA Seq methodology consists of several key steps: RNA extraction, enrichment (poly A or rRNA), fragmentation and library construction (with or without barcodes/UMIs), massive sequencing, and bioinformatics pipeline for QC, alignment, quantification, and interpretation. New approaches to 3′ mRNA-Seq and early barcoding (BRB-seq) significantly reduce costs and optimize experimental throughput.
Laboratory preparation
1) High-quality RNA extraction
- Strict control of RNA integrity (RIN ≥ 7 or equivalent), elimination of genomic DNA.
2) mRNA enrichment
- Conventional method: poly-A selection or rRNA depletion
- Recent alternatives: combined rRNA + poly A removal strategy adapted to dual RNA seq protocols (Ura et al., 2022, Shilpha et al., 2024).
3) Fragmentation/tagmentation
- Three approaches: enzymatic, chemical fragmentation or tagmentation via Tn5 transposase, which combines fragmentation and adapter insertion in a single step.
- Index barcodes added during the RT step for massive multiplexing.
4) cDNA synthesis and index/UMI addition
- Barcoding from reverse transcription, combined with tagmentation for high-throughput automation
5) PCR amplification & final preparation
- Addition of Illumina/Element Biosciences/MGI-compatible adapters.
- Size selection (~200 bp) and quantification.
High-throughput sequencing
- Use of NovaSeq/NextSeq (Illumina), Aviti (Element Biosciences) or DNBseq (MGI) platforms: millions of reads per sample.
- Adjustable depth: ~10–50 million reads for general quantification, >100 million for isoforms or rare RNAs.
- Multiplexing of several hundred samples via index/barcodes.
Bioinformatics pipeline
- Data quality: control with FastQC, adapter trimming, and low-quality sequences.
- Alignment & quantification: classic alignment or rapid pseudo-alignments. Quantification based on featureCounts, HTSeq, or UMI counting according to protocol.
- Differential expression analysis: normalization and statistical detection of differentially expressed genes
- Advanced steps: isoforms, alternative splicing, functional enrichment (GO/KEGG), visualization (heatmaps, PCA) and multi-omic integration (Georgakopoulos-Soares et al., 2022).
Georgakopoulos-Soares, I., Chan, C.S.Y., Ahituv, N. et al. High-throughput techniques enable advances in the roles of DNA and RNA secondary structures in transcriptional and post-transcriptional gene regulation. Genome Biol 23, 159 (2022). https://doi.org/10.1186/s13059-022-02727-6
Hasan, M & Feugang, Jean & Liao, Shengfa. (2019). A Nutrigenomics Approach Using RNA Sequencing Technology to Study Nutrient-Gene Interactions in Agricultural Animals. Current Developments in Nutrition. 3. 10.1093/cdn/nzz082. Figure
Shilpha, J., Lee, J., Kwon, JS. et al. An improved bacterial mRNA enrichment strategy in dual RNA sequencing to unveil the dynamics of plant-bacterial interactions. Plant Methods 20, 99 (2024). https://doi.org/10.1186/s13007-024-01227-x
Ura H, Togi S & Niida Y. A comparison of mRNA sequencing (RNA‑Seq) library preparation methods for transcriptome analysis. BMC Genomics 23, 303 (2022). (link.springer.com) 10.1186/s12864-022-08543-3