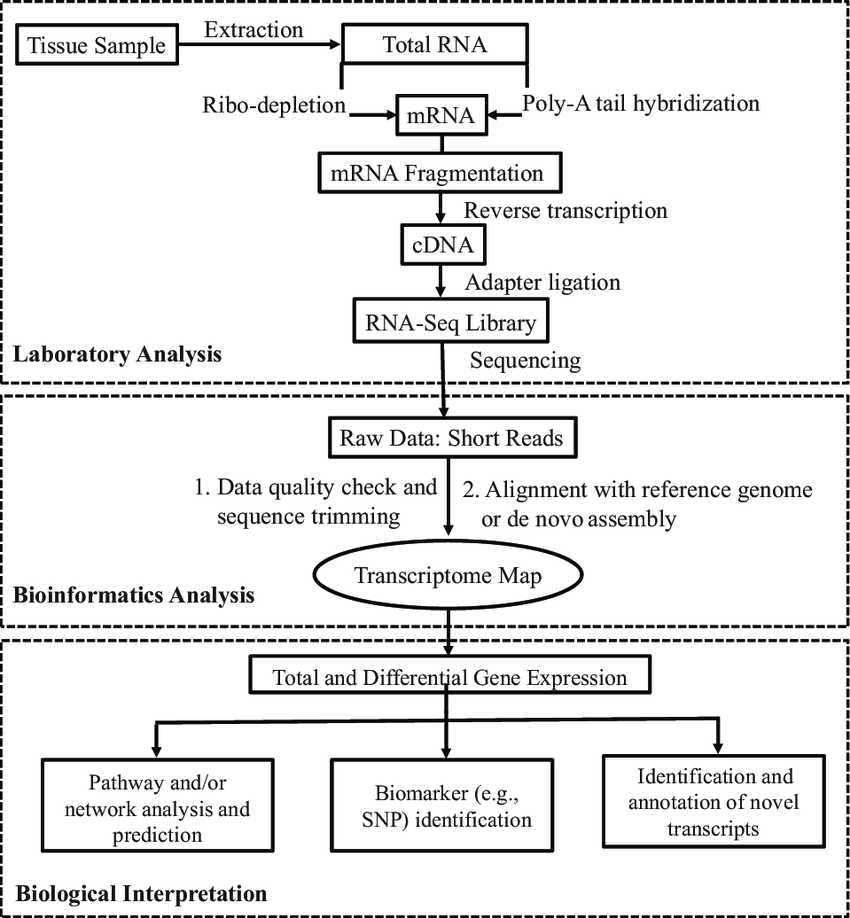
Le séquençage RNA‑Seq
Le séquençage RNA‑Seq à haut débit s’articule autour de plusieurs étapes clés : extraction d’ARN, enrichissement (poly‑A ou rRNA), fragmentation et construction de bibliothèque (avec ou sans barcodes/UMIs), séquençage massif, et pipeline bioinformatique de QC, alignement, quantification et interprétation. Les nouvelles approches de 3′‑mRNA‑Seq et de barcoding précoce (BRB‑seq) réduisent considérablement les coûts et optimisent le débit expérimental.
I) Préparation en laboratoire
1) Extraction d’ARN de haute qualité
Contrôle rigoureux de l’intégrité de l’ARN (RIN ≥ 7 ou équivalent), élimination de l’ADN génomique.
2) Enrichissement de l’ARNm
- Méthode classique : sélection poly-A ou déplétion rRNA
- Alternatives récentes : stratégie combinée d’élimination du rRNA + poly‑A adaptée aux protocoles dual RNA‑seq (Ura et al., 2022, Shilpha et al., 2024).
3) Fragmentation / tagmentation
-
- Trois approches : fragmentation enzymatique, chimique ou tagmentation via transposase Tn5 qui combine fragmentation et insertion d’adaptateurs en une seule étape
- Index-barcodes ajoutés dès l’étape de RT pour multiplexage massif.
4) Synthèse d’ADNc et ajout d’index/UMIs
-
- Barcoding dès la transcription inverse, combinée à la tagmentation pour automatisation à haut débit
5) Amplification PCR & préparation finale
-
- Ajout d’adaptateurs compatibles Illumina/Element Biosciences/MGI.
- Sélection de taille (~200 bp) et quantification.
II) Séquençage à haut débit
- Utilisation de plateformes NovaSeq/NextSeq (Illumina), Aviti (Element Biosciences) ou DNBseq (MGI) : millions de lectures par échantillon.
- Profondeur modulable : ~10–50 M lectures pour quantification générale, >100 M pour isoformes ou ARNs rares.
- Multiplexage à plusieurs centaines d’échantillons via index/barcodes.
III) Pipeline bio-informatique
- Qualité des données : contrôle avec FastQC, trimming adaptateurs et séquences de faible qualité.
- Alignement & quantification : alignement classique, ou pseudo-alignements rapides. Quantification basée sur featureCounts, HTSeq ou sur comptage UMI selon protocole.
- Analyse d’expression différentielle : normalisation et détection statistique des gènes différentiellement exprimés
- Étapes avancées : isoformes, splicing alternatif, enrichissements fonctionnels (GO/KEGG), visualisation (heatmaps, PCA) et intégration multi‑omique (Georgakopoulos-Soares et al., 2022).
Georgakopoulos-Soares, I., Chan, C.S.Y., Ahituv, N. et al. High-throughput techniques enable advances in the roles of DNA and RNA secondary structures in transcriptional and post-transcriptional gene regulation. Genome Biol 23, 159 (2022). https://doi.org/10.1186/s13059-022-02727-6
Hasan, M & Feugang, Jean & Liao, Shengfa. (2019). A Nutrigenomics Approach Using RNA Sequencing Technology to Study Nutrient-Gene Interactions in Agricultural Animals. Current Developments in Nutrition. 3. 10.1093/cdn/nzz082. Figure
Shilpha, J., Lee, J., Kwon, JS. et al. An improved bacterial mRNA enrichment strategy in dual RNA sequencing to unveil the dynamics of plant-bacterial interactions. Plant Methods 20, 99 (2024). https://doi.org/10.1186/s13007-024-01227-x
Ura H, Togi S & Niida Y. A comparison of mRNA sequencing (RNA‑Seq) library preparation methods for transcriptome analysis. BMC Genomics 23, 303 (2022). (link.springer.com) 10.1186/s12864-022-08543-3